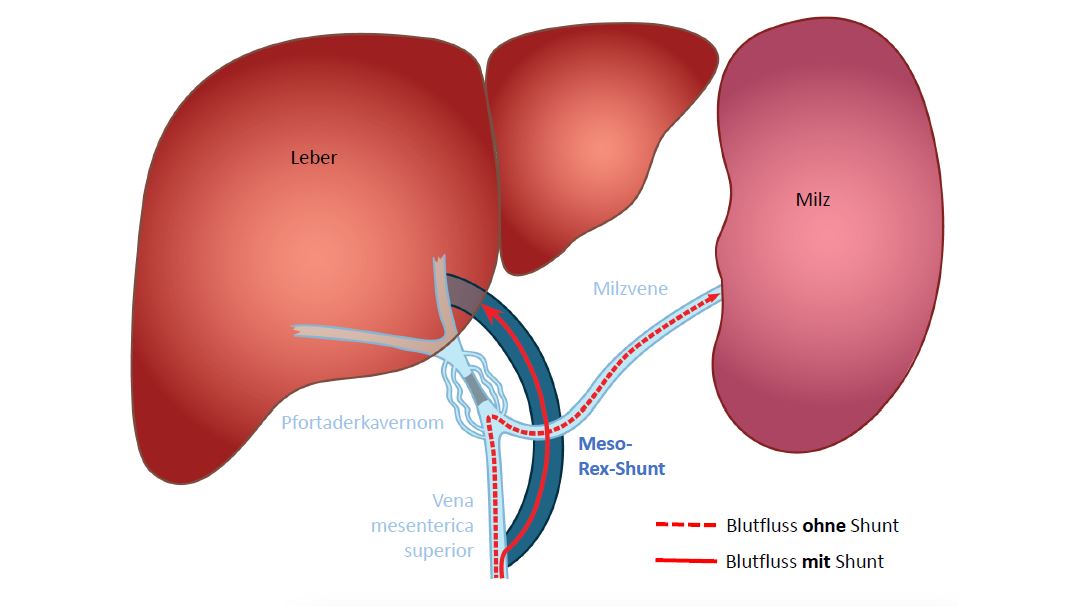
Die Leber erzeugt Enzyme und speichert diese. Die Nahrung, die wir essen, wird von diesen Enzymen in kleine Moleküle, die dem Organismus als Kraftstoff dienen,„aufgespalten“(abgebaut).
Ein Mensch, der an einer Stoffwechselerkrankung leidet, kann ein Enzym nicht oder nicht in ausreichender Menge produzieren. Bestimmte Nährstoffe, die nicht aufgespalten werden, sammeln sich im Körper und vergiften ihn.
Es gibt mehrere Stoffwechselerkrankungen: Diese können den Proteinstoffwechsel (Tyrosinämie, Phenylketonurie, Ahornsirupkrankheit), den Zuckerstoffwechsel (Glykogenose, hereditäre Fructoseintoleranz, Galactosämie) und den Fettstoffwechsel betreffen.
Sie können auch die Prozesse betreffen, die im Inneren der Zelle selbst ablaufen. Man spricht dann von mitochondrialen Erkrankungen oder lysosomalen Krankheiten.
Insgesamt treten Stoffwechselerkrankungen bei einem von 800 bis 2500 Neugeborenen auf.